HAEMOPHILIA - AN INSIGHT AND THE MOLECULAR BASIS OF THE DISORDER:
INTRODUCTION:
Haemophilia is a human genetic disorder caused by a sex-linked recessive allele (Campbell, Reece and Meyers, 2006). The disorders defined by the absence or deficiency of one or more of the proteins required for blood clotting - factor VIII and factor IX. (Campbell, Reece and Meyers, 2006). The lack of factor VIII causes classical Haemophilia (Haemophilia A), which accounts for approximately 83% of cases worldwide (Saladin, 2004); whereas the lack of factor IX - causing Haemophilia B, which accounts for approximately 15% of cases worldwide (Saladin, 2004). The prevalence of Haemophilia A is roughly one in ten thousand and in Haemophilia B, also known as Christmas disease, has a prevalence of one in fifty thousand. (Cahill, and Colvin. 1997). The affected individuals develop a variable phenotype of hemorrhage into the joints and muscles (Rumena, Stoian, Ivo. 2004). This causes excruciating pain and eventual joint immobility can result from the haematomas (masses of clotted blood in tissue) that form. (Saladin, 2004).
image one - represents the sex-linked recessive pattern of inheritence.
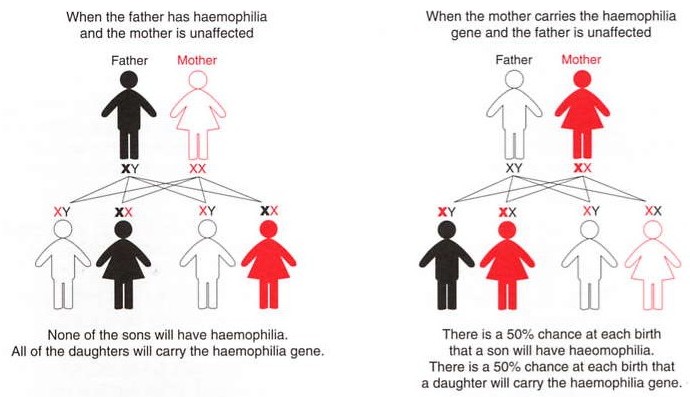
Reference - Haemophilia foundation Australia.
GENETICS:
Haemophilia A and Haemophilia B are inherited disorders in a recessive X-linked pattern (Rumena, Stoian, Ivo. 2004). Due to this X-linked pattern, Haemophilia is almost exclusively in males (Cahill, and Colvin. 1997). The sons of Haemophilia affected patients will be unaffected and the daughters will be carriers of the disorder; therefore the female carriers, with normal partners will result with half their sons as haemophiliacs and half their daughters as carriers (Cahill, and Colvin. 1997).
The genes for factor VIII and IX are located on the distal end of the long arm of the X chromosome, bands Xq28 and Xq27. (Cahill, and Colvin. 1997). Factor VIII (located in the subtelomeric Xq28 region) comprises of 26 exons and spans 186kb genomic DNA (Rumena, Stoian, Ivo. 2004). Factor IX (located at Xq27) has an entire sequence of 34kb and contains 8 exons, encoding 6 major domains of factor IX. (Giannelli. Et. Al. 1994). Factor VIII and factor IX are proteins which circulate as inactive precursors that are activated at the time of haemostatic challenge, via the instrinsic or extrinsic pathways of the coagulation cascade (Bowen. 2002). Factor VIII is a protein co-factor with no enzymatic activity, whereas factor IX is a serine protease with a requirement for factor VIII as a co-factor. (Bowen. 2002). Upon activation, and in the presence of calcium ions and phospholipid surfaces, factor VIII and factor IX form an active complex, which activates factor X. (Bowen. 2002). A deficiency or dysfunction of either factor VIII or factor IX compromises the activation of factor X and therefore ensuing steps of the coagulation cascade are also compromised resulting in fibrin deposition as either inefficient or non-existent and in turn, Haemophilia (Bowen. 2002).
CLASSIFICATION AND CLINICAL MANIFESTATIONS:
Children with severe haemophilia start bleeding at different ages of life; im one study, 44% of infants with haemophilia demonstrated their first bleeding episode during their first year of age. (Huether and McCance, 2004). During their first year, spontaneous bleeding is often minimal, however hematoma formation may result from injections and from firm holding ie. under the arms (Huether and McCance, 2004). By the age of 3-4, 90% of children with haemophilia have had episodes of persistant bleeding from relatively minor traumatic lacerations such as a cut to the lip or tounge (Huether and McCance, 2004). This is usually the first clinical manifestation of haemophilia (Huether and McCance, 2004).
Haemophilia is classified according to the level of clotting factors the patient has (Cahill, and Colvin. 1997). Its severity is varied and depends on the levels of clotting factors (Saladin, 2004). Half the amount of normal levels is enough to prevent symptoms (Saladin, 2004). Severely affected patients have <1% of normal factor levels, moderate patients with 1-4% and mild with 5-50%. (Cahill and Colvin. 1997). Smallcuts on the skin are not usually a problem (Campbell, Reece and Meyers, 2006) whereas during the 1980s intracranial bleeding was the leading cause of deaths for patients affected with Haemophilia, which proceeded by recorded trauma (Cahill, and Colvin. 1997). The majority of patients, severely affected with Haemophilia, treated before 1986 have been infected with HIV and many have since died of AIDS related illnesses (Cahill, and Colvin. 1997). This is the same scenario for hepatitis, for at that current time it was not known that the clotting factor concentrate used to treat the patients could transmit these viruses (Cahill, and Colvin. 1997). The development of inhibitory antibodies to factor VIII and factor IX poses another problem in management of Haemophilia (Cahill, and Colvin 1997). Patients whose Haemophilia is due to deletions or nonsense mutations are more likely to form inhibitors to either factor VIII or factor IX (Cahill and Colvin. 1997). The inhibitors have the ability to neutralise the activity of factor VIII or factor IX (Cahill, and Colvin. 1997).
MUTATIONS:
Point mutations, deletions, insertions and rearrangements/inversions are found in both factor VIII and factor IX genes (Bowen. 2002). Over 80 different mutations of factor VIII have been characterised and over 400 in factor IX. (Cahill, and Colvin. 1997). However, most of the mutations are private ie. they have been found in one or only a few unrelated families. (Antonarakis Et. Al. 1995). One form of mutation in which a very large sequence of DNA becomes inverted, has now been shown to account for almost all of the severe cases of Haemophilia A (Cahill,and Colvin. 1997). The inversion is mediated by the presence of three copies of a particular DNA sequence; one copy is located within inton 22 of the factor VIII gene and the other two are approximately 400kb telomeric to the first (Antonarakis Et. Al. 1995). Unequal crossing-over between two of these sequences results in the inversion of a portion of the factor VIII gene (exon 1-22) and therefore no intact factor VIII protein is produced (Antonarakis Et. Al. 1995). There are two types of inversions, a cross-over of the most distal sequence A and its IVS22 homolog which results with type one, and cross-over of the proximal extrogenic sequence A and its IVS22 homolog resulting with type two (Antonarakis Et. Al. 1995). When this cross-over occurs introns 1-22 are moved and their origin is inverted (Bowen. 2002). Antonarakis Et. Al. showed the analysis of 2093 severely affected Haemophilia A patients resulted with 35% withtype one and only 7% with type two. (Antonarakis Et. Al. 1995).
Deletions and nonsense mutations both give rise to severe Haemophilia (Bowen. 2002). Nonsense mutations occur at CGA codons where the CG transitions can give rise to missense or nonsense mutations, depending upon the codon in which the CG site resides: C to T transition is particularly damaging at this codon for it results with TGA - a stop codon (Bowen. 2002). This mutation compromises the activation of cleavage at that site, however partial activity may remain (Bowen. 2002).
TREATMENT:
Initially, fresh frozen plasma was the only product available for treatment; however with chromatographic purification, intermediate purity concentrations of factor VIII and factor IX became mainstay of treatment of factor VIII and factor IX deficiency throughout the 1970s and 1980s (Cahill and Colvin. 1997). The recent cloning of the factor VIII gene and the purification of the recombinatant factor VIII has resulted in factor VIII products that minimise the risk of transmission of viral infection ie. HIV/Hepatitis (Huether and McCance, 2004). unfortunately, those individuals with haemophilia who were treated before the current purficiation techniques, may have been exposed to HIV; its currently estimated that half of the 20,000 haemophiliacs in the United States have contracted HIV from blood products (Huether and McCance, 2004).
Therapy is based on the the replacement of factor VIII and factor IX to haemostatically adequate levels for the prevention or treatment of bleeds (Giuffrida Et. Al. 2008). Trials in gene therapy have been done in the goal to replace the defective gene sequence with a corrected version to eliminate disease for the lifetime of the patient (Murphy, and High. 2008). Progress is evident but the challenging task has not been accomplished at such time (Murphy, and High. 2008). Haemophilia A and B are among the most extensively researched diseases in the gene therapy field (Murphy, and High. 2008). A combination of factors including prevalence of disease, width of therapeutic window, ability to accommodate the corrected gene sequence in a gene transfer vector, reliability and availability of animal models of the disease and funding and support from disease-specific foundations, all contribute to the development of gene therapy in this disease (Murphy, and High. 2008).
Currently laboratory tests are of primary value in the evaluation of haemorrhagic disorders. (Huether and McCance, 2004). The three phases of coagulation can be individually assessed by simple, relilable tests; this can be seen in table one (Huether and McCance, 2004).
Table one
TEST
|
PHASE |
SIGNIFICANCE |
| Thrombin time |
III |
Measures fibrinogen; usually evelated first because without fibrinogen blood cannot clot |
| Prothrombin |
II |
a decrease indicates a deficency of factors II, V, VII, and X. Also used to monitor warfarin sodium therapy
|
| Activated partial thromboplastin time (ptt or aptt) |
I |
Assesses for factors XII, XI, IX, and VIII, also used to monitor heparin therapy |
Table one represents the the laboratory tests currently used in evaluation of haemorrhagic disorders (Huether and McCance, 2004).
ONLINE JOURNALS:
- Antonarakis, S., Rossiter, J.P., Young, M., Horst, J. Et. Al. 1995. factor VIII gene inversions in severe Haemophilia A: results of an international consortium study. American society of hematology, blood - vol 86, No. 6 pp 2206-2212.
http://bloodjournal.hematologylibrary.org/cgi/reprint/86/6/2206
- Bowen, D. J. 2002. Haemophilia A and Haemophilia B: molecular insights. Journal of Clinical Pathology, February; 55(1): 1–18
http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=1187139
- Cahill M.R., Colvin B.T. 1997. Haemophilia. Haemophilia Comprehensive Care Centre, Royal London Hospital, Whitechapel, UK.
http://pmj.bmj.com/cgi/content/abstract/73/858/201
- Giannelli, F., Green, P.M., Sommer, S.S., Et. Al. 1994. Haemophilia B: database of point mutations and short additions and deletions, fifth edition. nucleic acids research vol. 22 No. 17
http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=308314&log$=activity
- Giuffrida, A., Genesini, S., Franchini, M., Gironcoli, M., Aprili, G., Gandini, G. 2008. Inhibitors in mild/moderate haemophilia A: two case reports and a literature review. SIMTI Servizi Srl, July; 6(3):163-168
http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2626868
- Mannucci, P.M 2008. Autoimmune haemophilia. SIMTI Servizi Srl. January; 6(1): 6-7.
http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2626853
- Murphy, S. and High, K. 2008. Gene therapy for Haemophilia. The Authors Journal compilation Blackwell Publishing Ltd, March; 140(5): 479–487
http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2408641
ONLINE RESOURCE:
BOOKS:
- Campbell, N., Reece, J., Meyers, N. 2006. Biology. 7th Ed. Pearson Benjamin Cummings. Australia
- Huether, S.E., McCance, K.L. 2004. Understanding Pathophysiology 3rd Ed. Mosby. Salt lake city, Utah.
- Saladin, K. 2004.Anatomy and physiology - the unity of form and function. 3rd Ed. McGraw-Hill. New York. NY.
{kind=link}
Comments (0)
You don't have permission to comment on this page.